In short time so much data has been collected that what was before a largely empty map is now one of the most complete maps of Haiti. Michael Maron posted some before and after images on Flickr:
Before the earthquake (Jan 12th) :
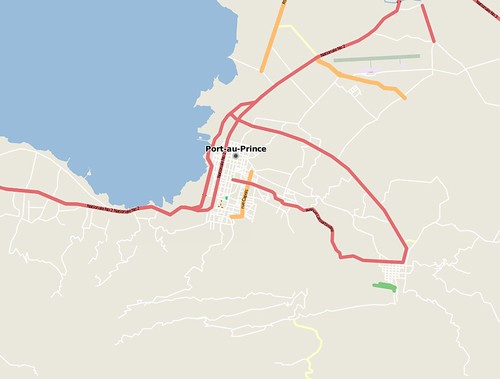
and by Jan 14th:
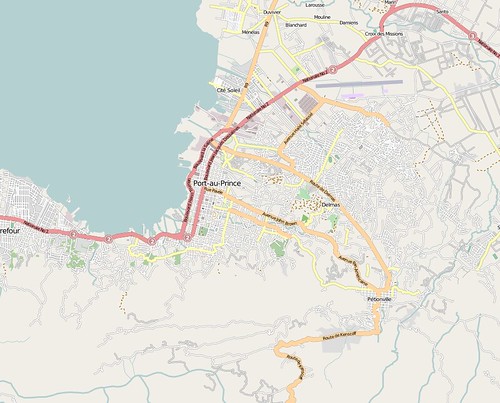
Beyond just marking roads, there is also an analysis of damaged buildings and displacement camps. This rendering, taken from the OSM wiki, shows damaged buildings and refugee camps mapped within OpenStreetMap.

A few other geographical projects related to Haiti are described at crisiscommons.org .
Resources:
- Project Haiti at OpenStreetMap
- Haiti Crisis Map shows the tagging of damaged buildings and camps, also can overlay sattelite images

{kind=link}